A multi-institutional team supervised jointly by Sam Gandy, Ottavio Arancio, and Michelle Ehrlich, published in Alzheimer's Dement., turned a designed cyclic D,L-α-peptide into a conformational sensor for the soluble, non-fibrillar aggregates of amyloid beta, NFA-Aβ, that evade standard detection. With that peptide in hand, the investigators showed that these oligomers accumulate with age in a mouse brain that never forms plaques, degrade presynaptic transmission, and reduce mitochondrial energy production in neurons, all without provoking the inflammation long assumed to drive the disease.
Fibrillar amyloid has dominated both diagnosis and therapy, yet plaque burden correlates poorly with cognitive decline. Suspicion has shifted toward NFA-Aβ, the oligomers and protofibrils that slip past amyloid positron emission tomography, PET, and current fluid biomarkers. Studying them has been difficult, because most models flood the brain with synthetic peptides or bury the oligomers among fibrils. The APPE693Q, or Dutch, mouse offers a rare alternative: its mutation swaps a glutamine for a glutamic acid inside the Aβ sequence, disrupting fibril assembly and favoring endogenously generated oligomers without parenchymal plaques.
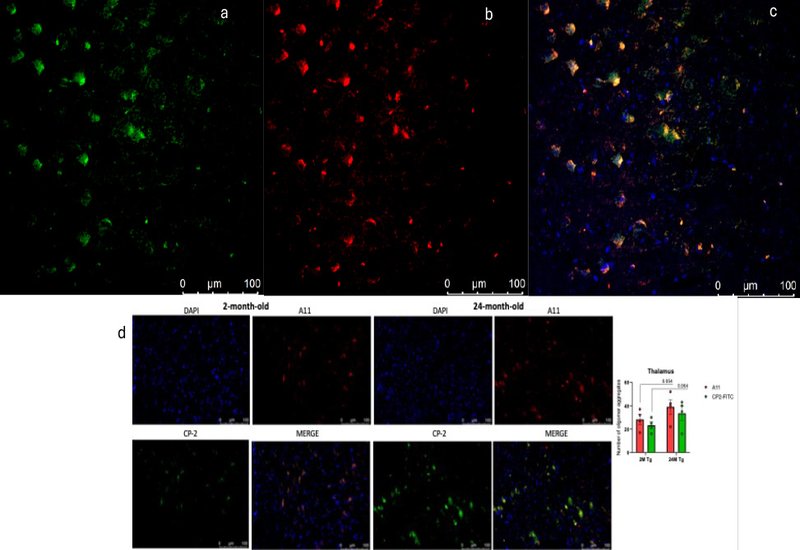
Seeing those oligomers required chemistry. The cyclic D,L-α-peptide at the center of the study, CP-2, was innovated in the laboratory of Shai Rahimipour at Bar-Ilan University and developed further by the lab of William Lubell at the Université de Montréal, with radiolabeling chemistry from Brigitte Guérin at the Université de Sherbrooke. CP-2 folds into a nanotube-like architecture that recognizes the prefibrillar oligomer epitope, binding the same conformation as the A11 antibody and stabilizing Aβ dimers and trimers. Conjugated to fluorescein, the peptide labeled endogenous NFA-Aβ across cortex, thalamus, and hippocampus, rising between 2 and 24 months of age while fibrillar markers stayed flat.
The functional consequences appeared at the synapse. Basal transmission and long-term potentiation remained intact, but aging Dutch mice developed selective defects in short-term presynaptic plasticity: weakened post-tetanic potentiation, faster synaptic fatigue, and altered refilling of the readily releasable vesicle pool. Single-cell RNA sequencing localized the strongest transcriptional disturbance to excitatory neurons, where genes for protein translation rose and genes for the electron transport chain fell. Electron microscopy and direct enzymology matched that signature: fewer and shorter mitochondria in CA1 presynaptic boutons, and reduced complex I activity by 12 months.
The team also reported what it did not find. Single-cell profiling detected no disease-associated microglia and no inflammatory transcriptional signature, even as the complement protein C1q increasingly tagged synapses for removal. The result reframes the amyloid cascade in these mice: neither fibrils nor neuroinflammation is required for the aging-related learning deficit. The clinical implication bears on the antibody era. Because lecanemab and donanemab slow decline only modestly, the authors argue that residual NFA-Aβ, invisible to fibril PET and fluid assays, may persist after treatment, and that clearing both fibrillar and non-fibrillar Aβ may be necessary for full benefit. A peptide-based tracer, the [64Cu]-NOTA cyclic D,L-α-peptide now in development, points toward monitoring that depletion directly in the living brain.
About the Contributors
The work divides along two axes. The peptide chemistry and molecular imaging that enabled it came from a transatlantic collaboration. The cyclic D,L-α-peptide ligand was designed and synthesized by Shai Rahimipour and Kuldeep Tripathi at Bar-Ilan University, whose laboratory leads the development of this peptide architecture. Brigitte Guérin at the Université de Sherbrooke developed the radiolabeled imaging chemistry. William Lubell at the Université de Montréal, a co-investigator on the supporting Cure Alzheimer's Fund award, contributed to manuscript review and editing. Charles Glabe at the University of California, Irvine supplied the A11 conformational antibody that anchored oligomer detection.
The neuroscience was led from Mount Sinai and Columbia. Emilie Castranio carried the study as first author. Merina Varghese and Dara Dickstein led the electron microscopy and synaptic ultrastructure; Elentina Argyrousi, Hong Zhang, and Ottavio Arancio ran the electrophysiology at Columbia; Minghui Wang and Bin Zhang performed the single-cell transcriptomics; Efrat Levy guided the NFA-Aβ biology. Linda Söderberg and Lars Lannfelt at BioArctic provided the protofibril assays, and Akiko Asada, Kanae Ando, and Toshiharu Suzuki at Tokyo Metropolitan University contributed the mitochondrial measurements. Sam Gandy, Ottavio Arancio, and Michelle Ehrlich shared senior supervision.